
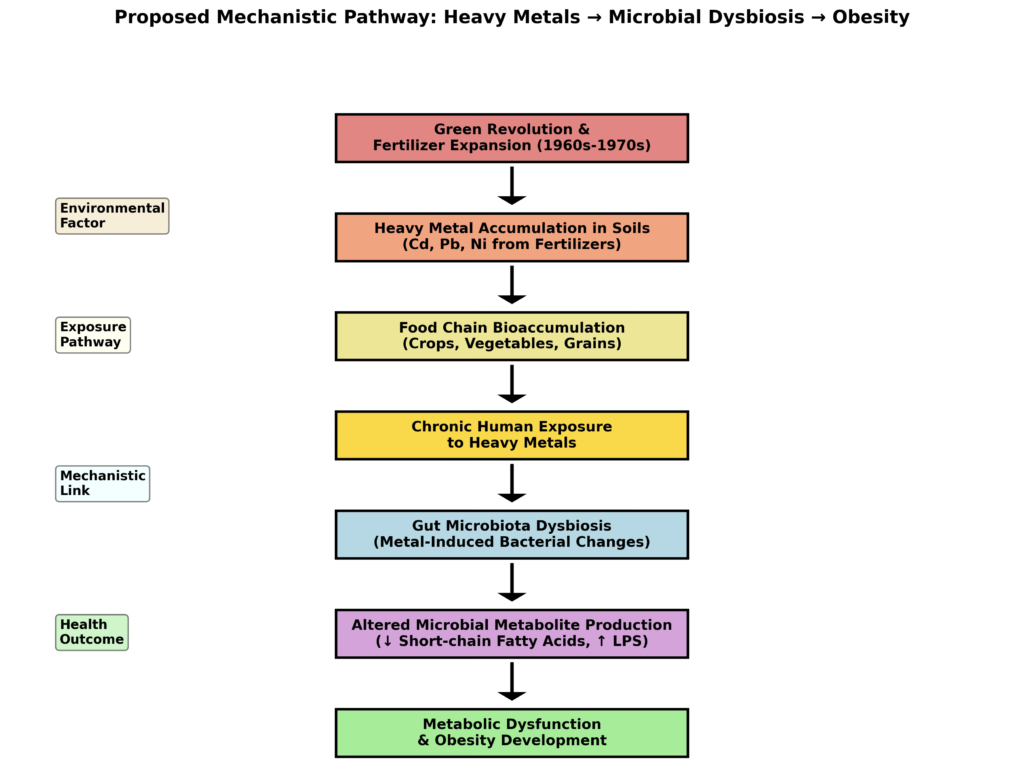
This mechanistic integrative review advances a falsifiable hypothesis to explain the rapid, population-wide emergence of the United States obesity epidemic beginning in the late 1970s. We propose that chronic dietary exposure to heavy metals, introduced at scale through agricultural intensification and phosphate fertilizer use, disrupted gut microbial metallomics and thereby primed the population for metabolic dysfunction. This framework addresses key epidemiological constraints by invoking a shared exposure capable of affecting all demographic groups within a short temporal window, without requiring genetic change or primary behavioral shifts. We synthesize evidence that cadmium, arsenic, lead, and nickel, and related metals function as metabolic toxicants and obesogens by impairing adipose biology, mitochondrial bioenergetics, redox homeostasis, and insulin signaling. Critically, these metals also impose selective pressure on the gut microbiota, enriching metal-tolerant taxa while depleting short-chain fatty acid–producing and barrier-supportive commensals.
The resulting dysbiosis disrupts microbial metal homeostasis, reduces fermentative signaling, compromises intestinal barrier integrity, promotes systemic inflammation, and converges on insulin resistance as a unifying pathophysiological outcome. In this model, heavy metal exposure acts as a permissive upstream factor that amplifies susceptibility to weight gain, while increased caloric availability and dietary change operate as proximate triggers. The hypothesis is testable through retrospective analysis of historical food and soil metal burdens, evaluation of temporal microbiome shifts consistent with metal-driven selection, and integration of metallomic biomarkers with longitudinal metabolic outcomes. By explicitly accounting for the timing, simultaneity, and population-level nature of the obesity epidemic, this microbial metallomics framework reframes obesity as environmentally induced metabolic inflexibility and identifies actionable targets for prevention at the food system and research levels.
Obesity epidemic, Heavy metals, Microbial metallomics, Gut microbiome, Environmental obesogens, Insulin resistance, Metabolic syndrome, Dysbiosis, Phosphate fertilizers, Food system contamination, Short-chain fatty acids, Intestinal barrier dysfunction, Environmental toxicology, Population-level metabolic disruption
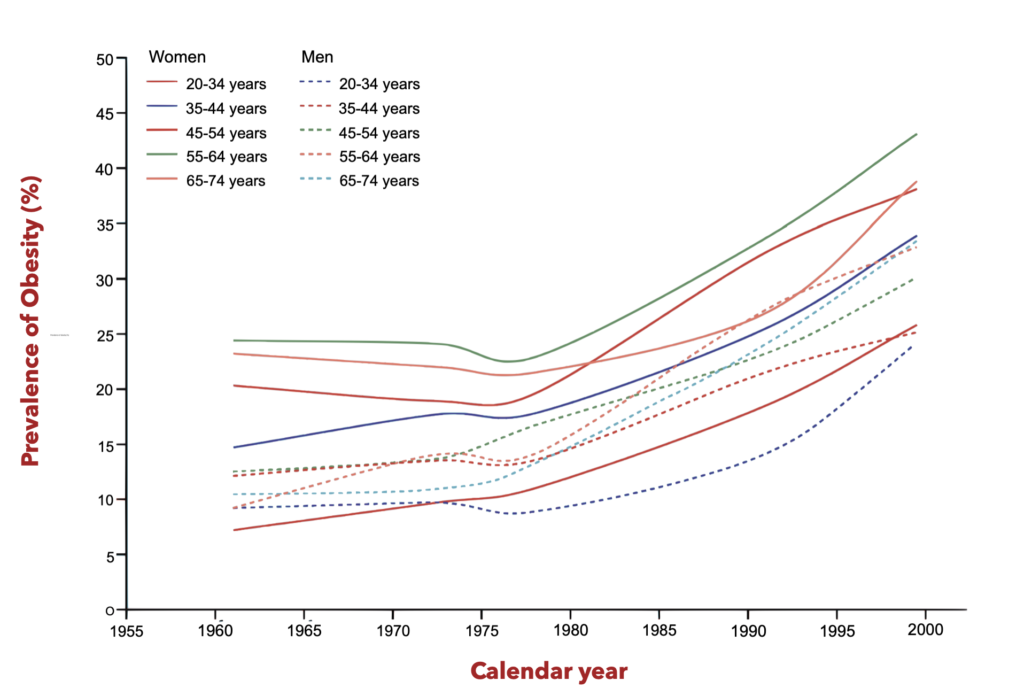
The emergence of the obesity epidemic in the United States presents a unique epidemiological puzzle that resists explanations centered on genetic factors or individual behavioral choices. As documented in the landmark analysis by Rodgers and colleagues [1], the obesity epidemic began in the late 1970s across the entire US population, affecting all age groups, sexes, and ethnic groups simultaneously This pattern of rapid, synchronized population-level change within a matter of years rather than decades explicitly rules out factors with long biological induction periods or factors that only affect certain subgroups.
The Rodgers analysis (Figure 1) demonstrates that the timing requirements for causality necessitate factors that achieved rapid, mass exposure in the late 1970s [1]. While the proposed agricultural explanation involving farm bill changes, high-fructose corn syrup, and increased food portion sizes remains plausible [1], an equally compelling but underexplored hypothesis deserves consideration: the widespread contamination of the food supply with heavy metals through agricultural intensification via urea and phosphate fertilizer use, operating through disruption of gut microbial metallomics to induce metabolic dysfunction and drive an obesity epidemic.
Epidemiological Association Between Heavy Metals and Metabolic Dysfunction
The epidemiological evidence linking heavy metal exposure to obesity and metabolic syndrome has accumulated substantially over the past two decades. Multiple cross-sectional studies utilizing large nationally representative datasets have established consistent associations between circulating and urinary heavy metal concentrations and increased body mass index, central obesity, and metabolic syndrome.
A comprehensive examination of data from the Korea National Health and Nutrition Examination Survey revealed that elevated concentrations of lead, mercury, cadmium, and nickel are associated with distinct metabolic health profiles, with certain metal exposure patterns correlating with heightened prevalence of obesity and diabetes [2]. Similarly, analysis of the US National Health and Nutrition Examination Survey (NHANES) data spanning 1999 to 2018 demonstrated that cadmium levels were positively associated with cardiovascular-kidney-metabolic dysfunction in a representative sample of American adults [3].
The association between individual heavy metals and obesity demonstrates dose-responsive patterns in some cases. In a large cross-sectional study of Chilean adults, exposure to arsenic showed association with obesity across most geographic areas, while mercury exposure was linked to metabolic syndrome, obesity, and cardiovascular conditions in central regions, suggesting both metal-specific and geographically-dependent metabolic effects [4]. For cadmium specifically, research indicates a bidirectional relationship with metabolic disease that varies with age, sex, and baseline metabolic status, with some studies showing U-shaped or threshold-dependent associations [5].
Adipose Tissue as a Primary Target of Heavy Metal Toxicity
Recent research has identified adipose tissue as a particularly vulnerable target for heavy metal-induced metabolic disruption. A comprehensive review of adipotropic effects demonstrated that mercury, cadmium, lead, and arsenic directly influence adipose tissue physiology through dose-dependent mechanisms [6]. In vitro studies reveal both upregulation and downregulation of adipogenesis associated with aberrant expression of key adipogenic pathways, namely CCAAT/enhancer-binding protein (C/EBP) and peroxisome proliferator-activated receptor gamma (PPAR).
The dose-dependency of these effects is particularly important: at low-dose exposures, heavy metals may stimulate adipogenesis, leading to increased adipose tissue accumulation, while higher doses inhibit adipocyte differentiation, reducing the lipid-storage capacity of adipose tissue and promoting ectopic lipid accumulation in liver and muscle [6]. This mechanism is mechanistically consistent with the paradoxical observation that metabolic dysfunction can occur in both obese and non-obese individuals exposed to heavy metals.
Experimental evidence in animal models demonstrates this principle directly. Gestational cadmium exposure in mice induces sex-specific hepatic insulin insensitivity, obesity, and metabolic syndrome in adult female offspring, establishing that developmental exposure to cadmium acts as a delayed-acting, sex-specific obesogen [7]. This developmental programming occurs through alterations in gene expression patterns related to oxidative stress, mitochondrial dysfunction, and retinoic acid signaling during critical developmental windows, findings that parallel the mechanisms of action of other developmental obesogens [8].
Agricultural intensification as a discrete historical exposure shift
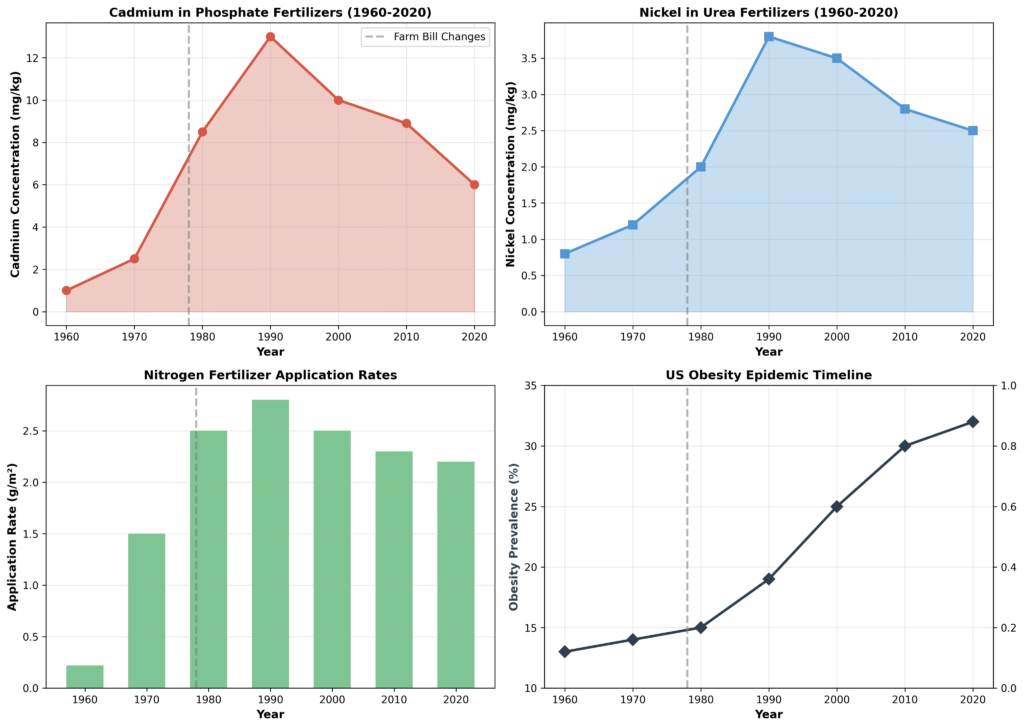
Prior to the mid-20th century, population-level exposure to cadmium and other heavy metals arose predominantly from localized industrial emissions and occupational settings [9]. This exposure pattern changed fundamentally with the post-1960 transition to intensive synthetic fertilizer use. A comprehensive historical reconstruction of fertilizer production, composition, and application from 1960–2025 demonstrates that agricultural inputs became the dominant, population-wide pathway for chronic dietary metal exposure, operating independently of individual behavior or geography [10].
Crucially, this transition is not gradual or diffuse. Nitrogen fertilizer application in the United States increased from approximately 0.22 g/m² in 1940 to 1.5 g/m² by the late 1970s, coinciding with the widespread adoption of synthetic urea as the dominant nitrogen source [1,10]. Phosphate fertilizer use expanded in parallel following the 1970s farm-bill–driven agricultural intensification [1]. These changes created a shared, rapid exposure pathway capable of affecting the entire population within a single decade.
Phosphate fertilizers and cadmium: a temporally constrained exposure pulse
Phosphate fertilizers derived from phosphate rock contain cadmium as an intrinsic contaminant. Historical analyses of fertilizer composition show that cadmium concentrations in commercial phosphate fertilizers increased steadily from the 1960s through the 1980s and peaked during the 1990s, with some products containing approximately 12–14 mg/kg cadmium, substantially exceeding both earlier background levels and modern regulatory thresholds [10].
Importantly, this peak contamination occurred after the initial expansion of fertilizer use but before the stabilization and regulatory tightening of the 2000s. Soil surveys summarized in the same historical analysis show that intensively fertilized agricultural soils accumulated cadmium to concentrations of approximately 0.8–1.5 mg/kg in North America, with higher levels in regions subjected to prolonged phosphate application [10]. Cadmium residence times in soil on the order of 20–40 years indicate that fertilizer-derived cadmium introduced during the 1970s–1990s would remain bioavailable well into subsequent decades [10].
Independent field studies corroborate this fertilizer-driven soil loading. Multivariate source-apportionment analyses in intensively cultivated tomato fields identified phosphate fertilizers as a dominant anthropogenic contributor to cadmium, arsenic, and lead enrichment in agricultural soils [11]. Similarly, sediment analyses near phosphate mining and application zones demonstrate downstream accumulation of fertilizer-derived metals in freshwater systems associated with rice cultivation [12].
Urea fertilizers and nickel: a parallel, previously underappreciated pathway
In parallel with phosphate expansion, urea fertilizers introduced a distinct nickel exposure channel that has been largely absent from obesity-focused environmental models. Nickel is an essential cofactor for plant urease activity, and low-level nickel presence was historically tolerated or even beneficial for crop nitrogen utilization. However, the historical fertilizer analysis shows that nickel concentrations in commercial urea increased from approximately 0.8 mg/kg in the 1960s, to 0.8-1.2 mg/kg above background levels in the 1970s, to 3.5–4.2 mg/kg during the 1990s and early 2000s, reflecting both industrial scaling and insufficient contaminant removal during fertilizer manufacture [10].
Critically, soil monitoring data indicate that nickel enrichment above background was already detectable in high-fertilizer-use regions by the late 1970s, with concentrations rising in parallel with nitrogen application maps [10]. Unlike cadmium, nickel exhibits moderate soil mobility under common agricultural pH conditions, facilitating uptake into crops, particularly legumes and leafy vegetables [10]. Thus, urea fertilizers created a second, temporally aligned dietary metal exposure that intensified during the same window as phosphate-derived cadmium.
Food-chain propagation and population-wide exposure
Once introduced into agricultural soils, fertilizer-derived metals propagate efficiently through the food chain. Rice serves as a particularly important bioaccumulator of arsenic and cadmium, with inorganic arsenic species predominating and showing geographic variation correlated with fertilizer use and soil characteristics [13]. Beyond rice, wheat, vegetables, and animal feeds accumulate fertilizer-derived metals at concentrations that exceed health-based guidance values for vulnerable populations, including infants and pregnant women [13]. The soil-crop-animal transfer pathway results in cumulative exposure across trophic levels.
Analyses of agricultural food systems demonstrate that cadmium, arsenic, chromium, nickel, and lead increase progressively from soil to crops to animal products, creating a population-wide dietary exposure that is independent of individual behavior or industrial proximity [14].
Satisfying the epidemiological timing constraint
The fertilizer-mediated contamination pathway uniquely satisfies the epidemiological constraint identified by Rodgers and colleagues [1]. The historical record shows that both phosphate-derived cadmium and urea-derived nickel increased rapidly between the early 1970s and mid-1980s, reached maximal intensity in the 1990s, and persisted due to long soil residence times [10]. This exposure trajectory aligns precisely with the sudden, synchronized emergence of the U.S. obesity epidemic beginning in the late 1970s, affecting all age groups, sexes, and ethnicities simultaneously [1]. Unlike explanations requiring slow genetic change or subgroup-specific behaviors, fertilizer-driven heavy metal exposure represents a shared, time-locked environmental perturbation capable of exerting population-scale metabolic effects within years rather than generations.
Insulin Resistance as the Convergent Mechanism
Multiple mechanistic pathways through which heavy metals induce metabolic dysfunction converge on impaired insulin signaling and insulin resistance. Nickel exposure provides a particularly illustrative example: in a cross-sectional analysis of NHANES data, urinary nickel concentration was independently associated with metabolic dysfunction-associated steatotic liver disease, with mediation analysis revealing that insulin resistance mediated approximately 73.69% of the association between nickel exposure and MASLD [14]. This finding establishes insulin resistance as a central pathophysiological process linking environmental metal exposure to metabolic disease.
Heavy metals disrupt insulin signaling through multiple mechanisms operating at the cellular and systemic levels. At the cellular level, metals interfere with the normal function of antioxidant defense enzymes by binding to critical thiol (-SH) groups of enzymes such as superoxide dismutase (SOD), catalase, glutathione peroxidase (GPx), and glutathione reductase (GR) [15]. This interference with antioxidant enzyme function results in excessive production of reactive oxygen species (ROS) and sustained oxidative stress [15]. Cadmium, for example, can suppress the synthesis of nitric oxide radical (NO), resulting in altered vaso-relaxation and impaired blood pressure regulation, which subsequently contribute to insulin resistance and metabolic dysfunction [15].
Additionally, heavy metals can directly substitute for essential metal cofactors in protein-binding sites. Lead and cadmium, for instance, possess charge and ionic radius similar to essential metals such as iron, zinc, and calcium, allowing them to displace these essential metals from their natural metal-binding sites. When displaced redox-active metals (copper, iron, manganese)are released from their protective protein complexes, they catalyze the decomposition of hydrogen peroxide through the Fenton reaction, generating damaging hydroxyl radicals and perpetuating oxidative stress [15]. This mechanism is particularly relevant to hepatic insulin resistance, where metals accumulate to high concentrations over decades due to the liver’s role as a major site of metal accumulation [16].
Mitochondrial Dysfunction and Metabolic Flexibility Loss
Heavy metal-induced mitochondrial dysfunction represents a second major pathway contributing to insulin resistance and obesity. The liver and adipose tissue, the primary organs affected in metabolic syndrome, depend critically on functional mitochondria for energy production, lipid metabolism, and metabolic flexibility. Heavy metal exposure triggers mitochondrial dysfunction through multiple mechanisms, including interference with mitochondrial bioenergetics, disruption of mitochondrial biogenesis, and impairment of mitochondrial dynamics [17].
The impact of heavy metals on hepatic mitochondrial function has been particularly well-characterized. Research demonstrates that chronic exposure to heavy metals results in altered hepatic lipid homeostasis via perturbation of steatosis gene expression and lipid species abundances, with activation of hepatic anti-oxidant response pathways and disruption of imprinted gene networks that have been previously implicated in metabolic dysfunction-associated steatotic liver disease [18].
This suggests that heavy metals, through mitochondrial dysfunction and impaired lipid metabolism, directly promote the development of fatty liver disease, a central feature of the metabolic syndrome that characterizes the obesity epidemic.
Defining Microbial Metallomics in the Context of Obesity
Metallomics, the comprehensive study of metal-binding molecules and their interactions with biological systems, has recently been extended to include the study of metal-microbe interactions in the context of host health. Mammalian metallomics, an advanced interdisciplinary field, explores the dynamic roles of metal elements within biological systems through the lens of metal-binding proteins, free metal ions, and importantly, gut microorganisms [19]. The gut microbiota itself functions as a “metagenome,” encoding metabolic pathways that are not present in the human genome, including pathways for metal homeostasis, detoxification, and metabolism.
Recent studies combining 16S rRNA gene sequencing with elemental analysis have identified specific correlations between gut bacterial taxa and circulating metal concentrations. In a direct examination of relationships between heavy metal exposure, gut microbiome composition, and metabolic markers in obese versus lean individuals, several key findings emerged [20].
Obese individuals exhibited significantly elevated stool concentrations of cadmium, zinc, iron, manganese, and phosphorus, while showing reduced concentrations of barium, vanadium, tungsten, titanium, germanium, neodymium, and sulfur compared to normal-weight controls. Most importantly, specific microbial taxa showed strong correlations with metal concentrations: Bifidobacteriaceae abundance was negatively correlated with cadmium (r=0.6629, p=0.0051) and fasting blood glucose (r=0.61, p=0.021), suggesting a protective association [20].
Heavy Metal-Induced Dysbiosis and Loss of Protective Microbial Taxa
Exposure to heavy metals induces dysbiosis—alterations in the composition and function of the gut microbiota—through selective toxicity to sensitive bacterial species while favoring growth of metal-tolerant species. Tributyltin (TBT), an environmental pollutant chemically similar in its metabolic effects to heavy metals, provides an illustrative model. TBT exposure in rats reduced relative abundance of beneficial Bacteroides and increased harmful taxa including Enterococcus, Bilophila, and Anaerovorax, changes that were associated with altered liver inflammation and lipid metabolism abnormalities [21].
A similar pattern occurs with heavy metal exposure: differential metal toxicity across bacterial species creates a selective pressure that enriches for metal-tolerant species while eliminating or suppressing beneficial commensals that confer metabolic protection to the host. The loss of specific beneficial bacteria, such as Bifidobacterium species, which show inverse correlations with both cadmium and glucose dysregulation [20], represents a critical loss of metabolic protection. This pattern has been consistently observed in the literature examining obesity-associated dysbiosis more broadly[22], [23].
Microbial Metal Resistance Genes and Metabolic Reprogramming
Heavy metal exposure selects for bacteria carrying metal resistance determinants, including genes encoding metal efflux pumps, metal-binding proteins, and enzymatic detoxification systems. These genes, encoded on plasmids or integrated into chromosomes, allow metal-resistant bacteria to survive in heavy-metal-contaminated environments.
However, the fitness cost of maintaining these resistance determinants, combined with the metabolic rewiring necessary to activate metal resistance pathways, results in a fundamental reprogram of bacterial metabolism.
The antibiotic resistance gene pool, which overlaps substantially with heavy metal resistance genes in environmental bacteria [19], becomes enriched in the gut microbiota under heavy metal exposure. This shift toward metal-resistant, antibiotic-resistant bacteria has profound implications for host metabolism, as it represents a transition from a microbiota optimized for host metabolic health to a microbiota optimized for bacterial survival under toxic stress. Such a dysbiotic shift away from saccharolytic fermenting bacteria toward proteolytic fermenting bacteria results in altered production of bacterial metabolites, particularly short-chain fatty acids (SCFAs) [24], which are critical signaling molecules that regulate intestinal barrier integrity, systemic inflammation, and host energy metabolism.
The Critical Role of Microbial Short-Chain Fatty Acid Production
Short-chain fatty acids (SCFAs), particularly butyrate, propionate, and acetate, are produced by commensal bacteria through fermentation of dietary fiber and resistant starch. These bacterial fermentation products have emerged as critical signaling molecules that regulate host metabolism through multiple mechanisms: they serve as histone deacetylase (HDAC) inhibitors that alter gene expression in intestinal epithelial cells and immune cells, they activate G-protein coupled receptors including GPR41 and GPR43 that regulate appetite and energy expenditure, and they provide approximately 10% of the host’s daily caloric intake [24].
Heavy metal-induced dysbiosis with loss of butyrate-producing bacteria such as Roseburia species, Faecalibacterium prausnitzii, and Bifidobacterium species results in a marked reduction in fecal and cecal SCFA concentrations. This reduction in butyrate production has profound metabolic consequences: it impairs intestinal barrier integrity through loss of tight junction protein expression (ZO-1, claudin, occludin), promotes intestinal permeability and bacterial lipopolysaccharide (LPS) translocation, triggers systemic inflammation through toll-like receptor 4 (TLR4) signaling, and directly reduces whole-body metabolic rate and increases susceptibility to weight gain [24].
Metallotolerant Bacteria and Altered Amino Acid Metabolism
A second critical pathway through which metal-resistant dysbiotic microbiota promote metabolic dysfunction involves alterations in amino acid metabolism, particularly tryptophan metabolism. Tryptophan, an essential amino acid required for protein synthesis, also serves as a precursor for serotonin synthesis (which regulates appetite and mood), the aryl hydrocarbon receptor (AhR) ligands (which regulate intestinal barrier function and immune tolerance), and kynurenine pathway metabolites (which regulate systemic metabolism and energy expenditure) [25].
Under conditions of dysbiosis induced by heavy metal exposure, the balance between bacterial tryptophan degradation pathways shifts dramatically. While some dysbiotic bacteria increase tryptophan catabolism through the kynurenine pathway, they simultaneously reduce the production of protective AhR ligands such as indole and indole derivatives.
This shift results in reduced AhR signaling in intestinal epithelial cells and type-3 innate lymphoid cells, impairing intestinal barrier function and promoting low-grade systemic inflammation [25]. The systemic inflammation resulting from dysbiosis and microbial lipopolysaccharide translocation contributes further to insulin resistance and metabolic dysfunction.
Population-Level Exposure Through the Agricultural System
The critical advantage of the heavy metal dysbiosis hypothesis is its perfect alignment with the epidemiological requirements established by Rodgers and colleagues. Heavy metals entering the agricultural system through phosphate fertilizers would affect the entire population simultaneously through the shared food supply, without requiring changes in individual genetics, behavior, or motivation.
The population-wide shift in food production driven by 1970s farm bill changes [1] would have rapidly increased fertilizer application rates across American agriculture. Within 5-10 years, this would have resulted in measurable accumulation of metals in soils and subsequent bioaccumulation in crops and animal products available in grocery stores across the nation.
Importantly, microbiome-mediated effects do not need to scale linearly with environmental metal concentrations, because relatively modest increases in dietary metal burden can cross ecological selection thresholds that shift community composition and function disproportionately. Unlike factors with long biological induction periods (such as intrauterine exposures with 60-70 year lags), heavy metal-induced dysbiosis could plausibly develop within months to a few years of sustained dietary exposure. The dysbiotic shift would occur most rapidly in the population subgroups with the highest dietary metal exposure, but would progressively affect all demographic groups consuming contaminated food [20].
Dysbiosis-Induced Metabolic Dysfunction Without Genetic or Behavioral Change
The elegance of the heavy metal dysbiosis hypothesis lies in its explanation for how metabolic dysfunction could emerge rapidly in genetically similar populations without invoking genetic change, behavioral change, or hormonal change (beyond those secondary to dysbiosis).
A shift from a eubiotic to dysbiotic microbiota composition would be accompanied by coordinated functional changes that converge on insulin resistance and weight gain. Specifically, heavy metal–selected dysbiosis is expected to reduce short-chain fatty acid production, particularly butyrate, thereby weakening anti-inflammatory signaling and metabolic regulation [24]. In parallel, disruption of intestinal barrier integrity through reduced tight junction protein expression increases permeability and facilitates microbial product translocation [24]. This promotes lipopolysaccharide-driven endotoxemia and downstream toll-like receptor 4 activation, amplifying systemic inflammation and impairing insulin signaling [26].
Dysbiosis also plausibly perturbs tryptophan metabolism by reducing microbial generation of aryl hydrocarbon receptor ligands, weakening epithelial and immune barrier maintenance, and further reinforcing low-grade inflammation [25]. In addition, altered bile acid biotransformation can disrupt secondary bile acid pools and host FXR/TGR5 signaling, with consequences for glucose homeostasis, lipid handling, and energy expenditure [27]. Finally, a community shift away from saccharolytic fermentation toward proteolytic metabolism can increase amino acid fermentation and the generation of ammonia and other potentially harmful amines, further contributing to hepatic and systemic metabolic stress [21].
Each of these shifts independently contributes to insulin resistance, impaired metabolic flexibility, reduced whole-body metabolic rate, and increased appetite drive. Collectively, they would produce the synchronized population-level increase in body weight that characterizes the obesity epidemic [1].
The Protective Potential of Eubiotic Microbiota
Recent research demonstrates that retention of eubiotic microbiota composition is protective against weight gain even in the face of dietary challenges. When wild primate (douc) microbiota were transplanted into germ-free mice, these animals remained lean even when exposed to a high-fat diet, while mice receiving captive douc microbiota (dysbiotic) gained significantly more weight regardless of diet [23]. This elegant experiment demonstrates that microbiota composition, independent of host genetics or diet, directly modulates obesity susceptibility.
Furthermore, specific bacterial taxa have been demonstrated to have direct anti-obesity effects. Parabacteroides distasonis, which is depleted by calorie restriction and associated with weight regain after weight loss, can be restored by oral supplementation to prevent rapid post-diet weight regain [27]. Akkermansia muciniphila, a mucin-degrading bacterium that strengthens intestinal barrier integrity and improves metabolic parameters [27], is depleted in obesity. These and other specific commensals represent a sort of “microbial metabolic reserve” that protects against weight gain and metabolic dysfunction.
Under the heavy metal dysbiosis hypothesis, widespread heavy metal contamination of the food supply in the late 1970s would have depleted this microbial metabolic reserve across the entire population simultaneously, explaining the sudden, synchronized, population-level emergence of the obesity epidemic.
Heavy Metals as a Complementary Mechanism Rather Than Exclusive Cause
The heavy metal dysbiosis hypothesis does not necessarily exclude the previously proposed explanations for the obesity epidemic, including increased food portion sizes, increased high-fructose corn syrup consumption, and increased food availability. [1]. Rather, the heavy metal contamination hypothesis can be viewed as a complementary or synergistic mechanism that would amplify the metabolic effects of increased food energy availability.
A population with eubiotic microbiota has multiple lines of metabolic defense: maintained SCFA production that enhances satiety and reduces appetite, intact intestinal barrier function that prevents metabolic endotoxemia, and metabolic flexibility that allows upregulation of energy expenditure in response to caloric excess.
A population with dysbiotic microbiota induced by heavy metal exposure would lack these defenses, rendering it exquisitely susceptible to weight gain when exposed to increased food availability and portion sizes. In this framework, the heavy metals would constitute a permissive factor that “primed” the population for obesity, while the dietary changes (such as increased calories, high-fructose corn syrup) would constitute the triggering factor that precipitated the epidemic.
A population with eubiotic microbiota has multiple lines of metabolic defense: maintained SCFA production that enhances satiety and reduces appetite, intact intestinal barrier function that prevents metabolic endotoxemia, and metabolic flexibility that allows upregulation of energy expenditure in response to caloric excess.
A population with dysbiotic microbiota induced by heavy metal exposure would lack these defenses, rendering it exquisitely susceptible to weight gain when exposed to increased food availability and portion sizes. In this framework, the heavy metals would constitute a permissive factor that “primed” the population for obesity, while the dietary changes (such as increased calories, high-fructose corn syrup) would constitute the triggering factor that precipitated the epidemic.
The Dose-Response Relationship and Vulnerability Heterogeneity
The heavy metal dysbiosis hypothesis also explains observed heterogeneity in obesity prevalence across different demographic groups. Dietary metal exposure varies substantially based on food choices, agricultural practices in regions of food production, and occupational exposures. Groups consuming higher amounts of metal-contaminated staple foods (rice, wheat) or living in regions with higher background metal contamination (due to mining history or agricultural practices) would experience more severe dysbiosis and greater obesity prevalence. This variation could account for the geographic, socioeconomic, and ethnic variation in obesity prevalence observed in the US [4].
This review proposes microbial metallomics as a unifying mechanistic framework capable of explaining the timing, simultaneity, and population-level nature of the US obesity epidemic in a manner not adequately addressed by existing models. By integrating environmental toxicology with microbiome-mediated metabolic regulation, the framework reconciles epidemiological observations that challenge purely genetic, behavioral, or caloric explanations.
Most importantly, this hypothesis does not reject established contributors such as increased food availability or dietary composition, but instead reframes them as proximate triggers acting upon a metabolically primed population rendered vulnerable by chronic environmental metal exposure. In this context, heavy metals function as permissive upstream disruptors that erode metabolic resilience through sustained interference with host–microbe homeostasis.
The strength of this framework lies in its convergence of mechanistic plausibility across multiple biological scales. Heavy metals are established obesogens that impair insulin signaling, adipose tissue function, mitochondrial bioenergetics, and redox balance, yet these direct host effects alone are insufficient to account for the rapid synchronization of metabolic dysfunction across diverse populations.
Incorporation of gut microbial metallomics provides a biologically responsive intermediary capable of translating low-dose, chronic exposure into systemic metabolic consequences. Selective pressure favoring metal-tolerant taxa, coupled with depletion of short-chain fatty acid–producing and barrier-supportive commensals, offers a coherent explanation for the observed loss of metabolic flexibility, increased endotoxemia, and insulin resistance characteristic of obesity-associated dysbiosis. This model also clarifies why susceptibility varies across individuals and regions, reflecting heterogeneity in dietary metal exposure, agricultural practices, and baseline microbiome composition.
At the same time, several limitations warrant explicit consideration. Much of the epidemiological evidence linking heavy metals to obesity and metabolic syndrome remains observational, and direct longitudinal data linking historical dietary metal exposure to microbiome shifts are limited. Dose thresholds for microbiome disruption are not yet well defined, and co-exposures to other environmental toxicants, socioeconomic factors, and dietary pattern changes may confound associations.
Accordingly, the present framework advances a causal hypothesis supported by coherence, temporality, biological plausibility, and consistency across disciplines, rather than definitive proof of causation. Causal confirmation will require longitudinal, interventional, and retrospective analyses capable of disentangling metal-specific effects from correlated environmental and dietary variables.
Crucially, the hypothesis generates testable predictions. Populations with lower dietary heavy metal exposure and similar diets to pre-1960s Western diets should retain a greater abundance of short-chain fatty acid–producing taxa and exhibit relative protection against metabolic dysfunction. Enrichment of metal-tolerant microbial taxa should precede overt insulin resistance and weight gain rather than emerge as a secondary consequence.
Interventions that reduce dietary metal burden or remediate contaminated soils should produce measurable microbiome recovery before or alongside metabolic improvement. Failure to observe these patterns would directly challenge this proposed framework.
The scope of this model is most immediately applicable to industrialized food systems characterized by intensive agriculture and fertilizer use, and it may not generalize to populations with minimal exposure to agricultural metal contamination. Both early-life and adult exposures are plausibly relevant, though developmental windows may confer heightened vulnerability through microbiome imprinting and metabolic programming. Extension of this framework beyond the US context will require region-specific exposure and microbiome data.
From a translational perspective, microbial metallomics highlights underappreciated intervention targets, including food system metal monitoring, fertilizer regulation, exposure threshold refinement, and microbiome-preserving agricultural practices.
Incorporating metallomic biomarkers into metabolic disease research may improve risk stratification and prevention strategies. More broadly, this framework reframes obesity as a disorder of environmentally induced metabolic fragility rather than simple caloric imbalance, positioning microbial metallomics as a critical mechanistic bridge between environmental toxicology, microbiome science, and chronic metabolic disease.
The heavy metal dysbiosis hypothesis provides a mechanistically plausible, epidemiologically consistent explanation for the sudden emergence of the obesity epidemic in the United States beginning in the late 1970s. The hypothesis satisfies the stringent requirements established by Rodgers and colleagues: it involves factors that achieved rapid, population-wide exposure without requiring genetic changes or individual behavioral modifications [1].
The convergence of evidence is compelling: (1) heavy metals, particularly cadmium and arsenic, are documented obesogens that disrupt adipose tissue function and induce insulin resistance [5],[6]; (2) phosphate fertilizers, the dominant modern source of heavy metal contamination in soils and food [9], became widely utilized during the exact period of obesity epidemic emergence; (3) heavy metals induce dysbiosis through selective toxicity to beneficial bacteria, favoring metal-resistant species and loss of SCFA-producing bacteria [20], [21]; (4) dysbiosis-induced loss of butyrate production, intestinal barrier disruption, and systemic inflammation directly cause insulin resistance and weight gain [24], [26]; (5) gut microbiota composition directly modulates susceptibility to weight gain independent of genetics or diet [23].
Future research should focus on retrospective analysis of archived food and soil samples from the 1970s-1980s period compared to contemporary samples to establish changes in heavy metal contamination of the food supply. Temporal analysis of shifts in gut microbiota composition in prospectively followed cohorts, with particular attention to loss of SCFA-producing bacteria, would test the microbial metallomics hypothesis directly.
Analysis of longitudinal microbiota compositional data in relation to metal exposure biomarkers would establish causal linkages. Such studies would represent a paradigm shift in understanding not merely the obesity epidemic, but more broadly how environmental toxicants shape the human microbiota to promote chronic metabolic disease.
[1] A. Rodgers, A. Woodward, B. Swinburn, and W. H. Dietz, “Prevalence trends tell us what did not precipitate the US obesity epidemic,” The Lancet Public Health, Apr. 2018, https://doi.org/10.1016/s2468-2667(18)30021-5
[2] S. Jeong and Y. Choi, “Investigating the Influence of Heavy Metals and Environmental Factors on Metabolic Syndrome Risk Based on Nutrient Intake: Machine Learning Analysis of Data from the Eighth Korea National Health and Nutrition Examination Survey (KNHANES),” Nutrients, Mar. 2024, https://doi.org/10.3390/nu16050724
[3] E. Agaba, T. L. Brown, and S. Adebamowo, “Abstract P174: Toxic Heavy Metals Are Associated With Increased Risk Of Cardiometabolic Disorders,” Circulation, Mar. 2022, https://doi.org/10.1161/circ.145.suppl_1.p174
[4] M. P, S.-P. A, and U. C, “Geographical variations in metal exposure and metabolic disorders in Chile using an exposome approach.,” Nov. 2025, https://doi.org/10.1038/s41598-025-23419-8
[5] S. S, “Is Environmental Cadmium Exposure Causally Related to Diabetes and Obesity?,” Dec. 2023, https://doi.org/10.3390/cells13010083
[6] A. Tinkov et al., “Adipotropic effects of heavy metals and their potential role in obesity,” Faculty Reviews, Mar. 2021, https://doi.org/10.12703/r/10-32
[7] T. W. Jackson, G. L. Ryherd, C. M. Scheibly, A. L. Sasser, T. Guillette, and S. M. Belcher, “Gestational Cd Exposure in the CD-1 Mouse Induces Sex-Specific Hepatic Insulin Insensitivity, Obesity, and Metabolic Syndrome in Adult Female Offspring,” Oxford University Press, Oct. 2020, doi: https://doi.org/10.1093/toxsci/kfaa154
[8] A. Kapama, C. Stefanaki, G. Mastorakos, and M. Papagianni, “The Role of Endocrine Disruptors in Childhood Obesity – Unraveling the Obesogens.,” Hormone Research in Paediatrics, Mar. 2025, https://doi.org/10.1159/000545043
[9] D. S, M. V, and S. G, “The Mechanisms of Cadmium Toxicity in Living Organisms.,” Nov. 2024, https://doi.org/10.3390/toxics12120875
[10] Pendergrass, K. “Heavy Metals, Microbial Metallomics, and the US Obesity Epidemic: A Mechanistic Examination of a Population-Level Metabolic Disruption.” Jan. 2026, https://doi.org/10.5281/zenodo.18439158
[11] N. Batapola et al., “Risk assessment of heavy metals in the freshwater lake sediments around Eppawala phosphate deposit, Sri Lanka,” Journal of the National Science Foundation of Sri Lanka, Jan. 2024, https://doi.org/10.4038/jnsfsr.v51i4.11473
[12] D. Zhao, P. Wang, and F.-J. Zhao, “Toxic Metals and Metalloids in Food: Current Status, Health Risks, and Mitigation Strategies,” Current Environmental Health Reports, Oct. 2024, https://doi.org/10.1007/s40572-024-00462-7
[13] A. H. Sani, A. Musa, and A. D. Musa, “Impact of Heavy Metals on the Environment and Human Health: A Comparative Analysis of Two Mining Communities in Niger State, Nigeria.,” International journal of research and scientific innovation, Dec. 2025, https://doi.org/10.51244/ijrsi.2025.12110111
[14] Z. Liu et al., “Insulin resistance as a mediator in the association between nickel exposure and metabolic dysfunction-associated steatotic liver disease,” Diabetology & Metabolic Syndrome, Jan. 2025, https://doi.org/10.1186/s13098-024-01567-7
[15] K. Jomov, S. Y. Alomar, E. Nepovimova, K. Kua, and M. Valko, “Heavy metals: toxicity and human health effects,” Archives of Toxicology, Nov. 2024, https://doi.org/10.1007/s00204-024-03903-2
[16] K. R et al., “Metals in the human liver: An underappreciated risk factor of hepatic insulin resistance and associated pathophysiology.,” Jul. 2025, https://doi.org/10.1016/j.envpol.2025.126844
[17] B. A. Lolescu et al., “Adipose tissue as target of environmental toxicants: focus on mitochondrial dysfunction and oxidative inflammation in metabolic dysfunction-associated steatotic liver disease,” Molecular and Cellular Biochemistry, Dec. 2024, https://doi.org/10.1007/s11010-024-05165-z
[18] L. Dameris, J. Hartsell, J. Chappel, X. Liu, and M. Cowley, “Cadmium exposure during adolescence and young adulthood induces signatures of metabolic dysfunction-associated steatotic liver disease,” Scientific Reports, Nov. 2025, https://doi.org/10.1038/s41598-025-22462-9
[19] T. X, T. Y, D. Y, Z. Q, H. C, and F. J, “Advances in Mammalian Metallomics: New Insights into Metal Dynamics and Biological Significance.,” Oct. 2025, https://doi.org/10.3390/ijms26199729
[20] A. B. M. Alamer, M. Komijani, and S. Shahrjerdi, “Heavy Metals, Gut Microbiota, and Biochemical Markers: Unraveling the Complexities of Obesity,” MicrobiologyOpen, Oct. 2025, https://doi.org/10.1002/mbo3.70071
[21] Y. Gh et al., “Gut microbiota-mediated tributyltin-induced metabolic disorder in rats.,” Nov. 2020, https://doi.org/10.1039/d0ra07502g [22] Q. Li et al., “Gut microbiota-derived 4-hydroxyphenylacetic acid (4-HPAA) inhibits weight gain and is negatively associated with childhood obesity,” Translational Pediatrics, Jun. 2025, https://doi.org/10.21037/tp-2025-158
[23] D. N. Sidiropoulos et al., “Wild primate microbiomes prevent weight gain in germ-free mice,” Animal Microbiome, May 2020, https://doi.org/10.1186/s42523-020-00033-9
[24] Z. M. Kundi et al., “Dietary Fiber from Oat and Rye Brans Ameliorate Western Diet-Induced Body Weight Gain and Hepatic Inflammation by the Modulation of Short Chain Fatty Acids, Bile Acids, and Tryptophan Metabolism.,” Molecular Nutrition & Food Research, Jun. 2020, https://doi.org/10.1002/mnfr.201900580
[25] P. B. Toft, H. Yashiro, D. Erion, M. Gillum, F. Bckhed, and T. Arora, “Microbial dietary protein metabolism regulates GLP1 mediated intestinal transit,” The FASEB Journal, Sep. 2023, https://doi.org/10.1096/fj.202300982R
[26] H. Liu, S. Bovornkitti, S. Soontornchai, X. Qiu, and W. Shi, “Mediating role of systemic inflammation in heavy metal-induced metabolic dysfunction-associated steatotic liver disease: Insights from NHANES 2017 to 2020,” Eurasian Journal of Medicine and Oncology, Nov. 2025, https://doi.org/10.36922/ejmo025330348
[27] M. Li et al., “Gut microbiota-bile acid crosstalk contributes to the rebound weight gain after calorie restriction in mice,” Nature Communications, Apr. 2022, https://
Pendergrass, K. (2026). Heavy Metals, Microbial Metallomics, and the US Obesity Epidemic: A Mechanistic Examination of a Population-Level Metabolic Disruption. Zenodo. https://doi.org/10.5281/zenodo.18434951
This work is licensed under a Creative Commons Attribution 4.0 International License.
 Heavy Metals in Fertilizers/ A Historical Analysis of Contamination Trends (1960-2025)
Heavy Metals in Fertilizers/ A Historical Analysis of Contamination Trends (1960-2025)
 Cadmium Effects in Infants and Children: A Comprehensive Review of Health Impacts, Microbiome Shifts, and Microbial Metallomics
Cadmium Effects in Infants and Children: A Comprehensive Review of Health Impacts, Microbiome Shifts, and Microbial Metallomics

